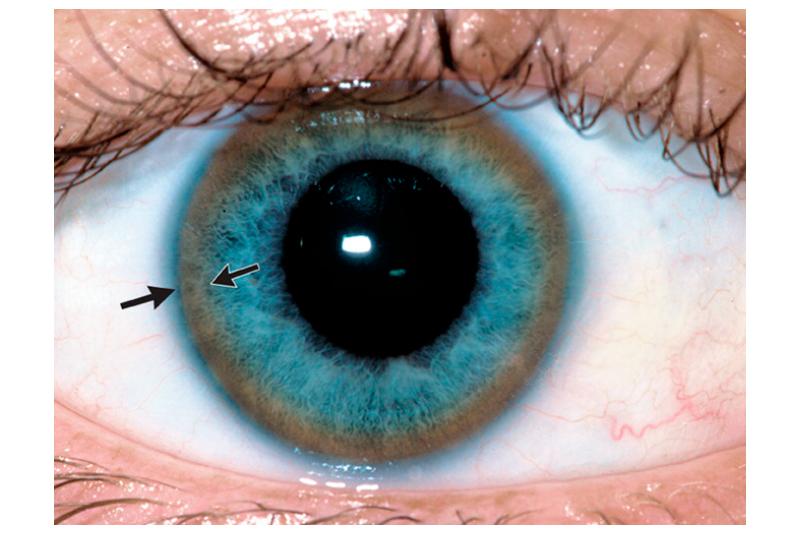
Следующим местом скопления меди можно назвать центральную нервную систему. Более всего страдают базальные ганглии головного мозга, провоцируя тяжёлые нейропсихические нарушения. Отложения меди видны и в роговице глаза — так называемое кольцо Кайзера-Флейшера. Медь поражает также сердце и суставы.
Болезнь Вильсона — это заболевание, передающееся по наследству, его причина заключается в нарушении накопления и транспорта меди в организме, вследствие чего происходит хроническая интоксикация. В крови понижается концентрация белка, который транспортирует медь, церрулоплазмина. Полное название болезни звучит как Болезнь Вильсона-Коновалова, гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля-Вильсона-Коновалова.
- Классификация болезни Вильсона
- Этиология и патогенез болезни Вильсона
- Клиническая картина болезни Вильсона
- Диагностика болезни Вильсона
- Лечение болезни Вильсона
- Прогноз при болезни Вильсона
- Профилактика болезни Вильсона
Патология провоцирует множественные наследственные заболевания внутренних органов и центральной нервной системы, особенно печени и промежуточного мозга. Передается это заболевание по аутосомно-рецессивному типу. Патологический ген ATP7B (находится в 13 хромосоме) мутирует, что и вызывает нарушения.
В случае, если человек получает дефектный ген от обоих родителей, болезнь начинает проявляться и прогрессировать. Человек, имеющий один патологический мутантный ген является носителем заболевания, концентрация меди отклонена от нормы, но нарушения незначительны.
Диагностирование заболевания приходится на возраст 10-13 лет. Болезнь проявляет себя как смешанный цирроз печени или почечная недостаточность. Чаще болеют мальчики. Распространённость болезни Вильноса повышается среди народностей, у которых популярны браки между кровными родственниками.
Заболевание встречается с частотой 1:30 000 населения. Аномальный ген, ответственный за развитие болезни, локализуется в области XIII хромосомы. Каждый больной является гомозиготным носителем этого гена. Заболевание распространено по всему миру, но чаще встречается среди евреев восточноевропейского происхождения, арабов, итальянцев, японцев, китайцев, индийцев и в популяциях, где часты близкородственные браки.
Причины болезни Вильсона-Коновалова
Гепатолентикулярная дегенерация является наследственным заболеванием и передается по аутосомно-рецессивному типу.
Заболевание встречается с частотой 1:30 000 населения. Аномальный ген, ответственный за развитие болезни, локализуется в области XIII хромосомы. Каждый больной является гомозиготным носителем этого гена. Заболевание распространено по всему миру, но чаще встречается среди евреев восточноевропейского происхождения, арабов, итальянцев, японцев, китайцев, индийцев и в популяциях, где часты близкородственные браки.
Гепатолентикулярная дегенерация вызвана мутацией гена переносчика меди, расположенного на 13-й хромосоме. Заболевание обычно проявляется на 2-3-м десятилетиях жизни преимущественно неврологическими и психическими симптомами либо симптомами, связанными с поражением печени (каждая из этих групп симптомов преобладает в трети случаев). Иногда симптомы появляются на первом десятилетии жизни, крайне редко — на шестом.
[1], [2], [3], [4], [5], [6]
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера. Биохимические исследования мочи обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
Лечение болезни Вильсона
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — леводопы, карбидопы, тригексифенидила.
Наиболее частой мутацией при БВК в славянской популяции является H1069Q, которая наблюдается в 30-50 % всех случаев в гомозиготном или (компаунд) гетерозиготном состоянии с другой генетической аберрацией гена ATP7B. Другими частыми аберрациями являются c.1340_1343del4, c.1770insT, c.2304insC, c.2532delA, с.3026_3028delTCA, 3029insT, 3031insC, с.3627_3630del4, 3649_3654del6, c.3942delAT, 3947delG.
БВК, гепатолентикулярная дегенерация, гепатоцеребральная дистрофия, болезнь Вестфаля — Вильсона.
Wilson disease, hepatolenticular degeneration.
Локализация гена на хромосоме
Полимеразная цепная реакция, фрагментный анализ.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Болезнь Вильсона — Коновалова (БВК) представляет собой наследственное аутосомно-рецессивное заболевание, вызываемое различными видами генетических аберраций в гене ATP7B. Данный ген кодирует белок медь-переносящую (транспортирующую) АТФ-азу, который участвует в метаболизме ионов меди. Мутации в гене ATP7B приводят к избыточному накоплению ионов меди в паренхиме печени, головного мозга и других органов с развитием структурно-функциональных нарушений органов и систем.
Наиболее частой мутацией при БВК в славянской популяции является H1069Q, которая наблюдается в 30-50 % всех случаев в гомозиготном или (компаунд) гетерозиготном состоянии с другой генетической аберрацией гена ATP7B. Другими частыми аберрациями являются c.1340_1343del4, c.1770insT, c.2304insC, c.2532delA, с.3026_3028delTCA, 3029insT, 3031insC, с.3627_3630del4, 3649_3654del6, c.3942delAT, 3947delG.
Для чего используется исследование?
- Диагностика болезни Вильсона — Коновалова
Когда назначается исследование?
- При подтверждении болезни Вильсона — Коновалова.
- При дифференциальной диагностике причин поражения печени, гиперферментемии и цирроза.
- При дифференциальной диагностике причин дистонии, гиперкинезов, экстрапирамидных нарушений.
Что означают результаты?
Обнаружение гомозиготной мутации или компаудной гетерозиготы подтверждает диагноз «болезнь Вильсона — Коновалова».
Обнаружение гетерозиготного носительства мутации в гене ATP7B не подтверждает диагноза «болезнь Вильсона — Коновалова» при отсутствии симптоматики.
При наличии характерных симптомов болезни Вильсона — Коновалова и гетерозиготного носительства аберраций в гене ATP7B вероятность достоверности диагноза «болезнь Вильсона — Коновалова» значительно повышается.
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды и индивидуальных генетических факторов.
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Вариант болезни Вильсона-Коновалова, при котором поражается печень, как правило, начинает развиваться у подростков только с одиннадцатилетнего возраста. Неврологическая форма обычно проявляется после девятнадцати лет, когда пубертатный период уже заканчивается, но у некоторых пациентов болезнь Вильсона-Коновалова развивается уже не в молодом, а в пенсионном либо предпенсионном возрасте.
Симптомы синдрома Вильсона-Коновалова
Вариант болезни Вильсона-Коновалова, при котором поражается печень, как правило, начинает развиваться у подростков только с одиннадцатилетнего возраста. Неврологическая форма обычно проявляется после девятнадцати лет, когда пубертатный период уже заканчивается, но у некоторых пациентов болезнь Вильсона-Коновалова развивается уже не в молодом, а в пенсионном либо предпенсионном возрасте.
Переизбыток меди в организме, характерный для синдрома Вильсона-Коновалова, вызывает такие сопутствующие заболевания, как сахарный диабет и цирроз печени, аневризму – патологических изменений сосудов, синдром Фанкони, проявления которого напоминают рахит, при этом атеросклероз развивается ускоренными темпами.
В чем же заключается лечение этой тяжелейшей болезни? Во-первых, это строгое соблюдение “печеночной” диеты (стол 5а), предполагающей исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые и др.). Однако основное лечение – постоянный прием препаратов, выводящих медь из организма. Главным из них является D-пеницилламин.

И.А. Иванова-Смоленская
профеcсор, доктор медицинских наук
ГУ НИИ неврологии РАМН
Открытый недавно ген болезни отвечает за синтез медь-транспортирующего белка (АТР7В). При гепатолентикулярной дегенерации обмен меди и медьсодержащих белков нарушается, появляется избыток “свободной” меди, которая в больших количествах откладывается в печени, мозге, роговице, а также выделяется с мочой. Не случайно диагностика болезни базируется на обнаружении характерных нарушений медного обмена. Благодаря идентификации гена в настоящее время возможна и ДНК- диагностика этого заболевания.
Поражение печени избытком “свободной” меди проявляется циррозом печени. Поражение мозга приводит к развитию тяжелой неврологической симптоматики: дрожанию конечностей и всего туловища, повышению мышечного тонуса, иногда сопровождающемуся болезненными спазмами, нарушением речи, глотания, снижению интеллекта. Отложение меди в роговице (по краю радужной оболочки) обусловливает формирование кольца Кайзера–Флейшера – буро-зеленоватого пигмента. По этому признаку диагноз болезни можно поставить безошибочно.
В чем же заключается лечение этой тяжелейшей болезни? Во-первых, это строгое соблюдение “печеночной” диеты (стол 5а), предполагающей исключение богатых медью продуктов (шоколад, кофе, орехи, бобовые и др.). Однако основное лечение – постоянный прием препаратов, выводящих медь из организма. Главным из них является D-пеницилламин.
Эти препараты назначаются по специальной схеме с постепенным увеличением дозы. К сожалению, в силу необходимости проведения пожизненного лечения и особых требований к химической чистоте препаратов отечественный аналог пеницилламина не может быть рекомендован при гепатолентикулярной дегенерации из-за высокой токсичности.
При длительном многолетнем приеме D-пеницилламина у некоторых больных гепатолентикулярной дегенерацией возникают побочные явления в виде дерматитов, анемии и иных осложнений. Поэтому был предложен альтернативный метод лечения солями цинка (оксид, сульфат и др.). Нами было предложено комбинированное лечение D-пеницилламином и препаратами цинка, что дает возможность снизить дозу и избежать побочных явлений. У больных в пресимптоматической стадии достаточно лечения только препаратами цинка.
Помимо этих методов, большое значение имеет гепатопротекторная терапия, направленная на максимальное улучшение функций печени.
Таким образом, при правильной терапии гепатолентикулярной дегенерации – тяжелейшего наследственного заболевания мозга и внутренних органов – в 80% случаев возможно клиническое выздоровление либо выраженное улучшение состояния больных при условии своевременной максимально ранней диагностики.
Страдающие болезнью Вильсона должны отказаться от алкоголя. Они не должны принимать препараты, токсичные для печени. Важно соблюдать диету, врачи назначают им стол 5а, и ограничивать поступление меди в организм. Из рациона следует исключить продукты с высоким содержанием меди в организме:
Болезнь Вильсона – это тяжелая наследственная патология обмена меди в организме, при которой медь откладывается во внутренних органах. В России эта болезнь известна как болезнь Вильсона-Коновалова.
Признаки
Неврологический вариант развивается позже, примерно в возрасте 19 лет. Обычно он начинается с неврологических симптомов – человеку становится трудно выполнять мелкие движения, появляются проблемы с речью, она замедляется и становится смазанной. Руки, ноги и туловище дрожат. Такие люди с трудом ходят, совершают много непроизвольных движений, они замедленны, мышечный тонус при этом повышен. Через некоторое время у страдающих болезнью Вильсона снижаются внимание и память, развивается депрессивное расстройство.
Характерный признак болезни Вильсона – кольцо Кайзера-Флейшера – зелено-бурый ободок по внешнему краю роговицы глаза. Он возникает из-за отложения меди в роговице.
Также при болезни Вильсона возможны поражения почек, миокарда, эндокринных желез, поджелудочной железы. Возможно и поражение суставов.
Описание
Это довольно редкое заболевание, им страдают 3 человека на 100 000 населения, причем в обществе, в котором распространены близкородственные браки, оно встречается чаще. Заболевание начинается в возрасте 11-25 лет. Однако это заболевание может развиться и у пожилых. Но при этом у них нет кольца Кайзера-Флейшера и неврологических симптомов. Мужчины страдают этим заболеванием чаще, чем женщины.
Причина заболевания – мутация в 13-й хромосоме. Именно там находится ген, отвечающий за правильный обмен меди в организме. Заболевание наследуется по аутосомно-рецессивному типу. То есть, для того, чтобы болезнь проявилась, ребенок должен получить по одному рецессивному (проявляющемуся только при наличии такого же парного гена) гену от родителей. Часто заболевание начинается после какого-либо толчка извне – вирусного гепатита, злоупотребления алкоголем, гепатотоксинов.
В организме взрослого человека содержится примерно 100-200 мг меди. Она участвует в процессе кроветворения, нормализует работу эндокринной системы, укрепляет стенки кровеносных сосудов, улучшает пищеварение, участвует в построении белков, укрепляет кости, обладает противомикробным и противовоспалительным действием, влияет на пигментацию кожи и волос. В сутки взрослому человеку необходимо 1-2 мг меди, беременным женщинам – 2-2,5 мг, детям, в зависимости от возраста, — от 1 до 3 мг.
Больше всего меди – в печени, почках, головном мозге и сердце. Примерно 90 % ее связано с церулоплазмином (белком плазмы крови). Выводится она с желчью. Однако если меди становится больше, чем белков, которые ее связывают, медь накапливается в печени. Это приводит к воспалению печени, позже к ее фиброзу, еще позже – к ее циррозу. Кроме того, несвязанная медь попадает в кровь и циркулирует по всему организму, откладываясь в различных органах – в сердце, роговице глаза, почках, головном мозге.
Из-за отложения меди в органах нарушается их работа. При отложении меди в сердце развивается кардиомиопатия, при отложении ее в почках развивается синдром Фанкони (нарушение функции почечных канальцев), при отложении в печени развивается цирроз печени или печеночная недостаточность, при отложении в мозге развиваются нейропсихические нарушения. Также последствиями неправильного метаболизма меди может быть нарушение толерантности к глюкозе (сахарный диабет), атеросклероз и расширение кровеносных сосудов (аневризма).
Диагностика
Для постановки правильного диагноза нужно пройти обследование у гастроэнтеролога и офтальмолога.
Также необходимо сделать общий и биохимический анализы крови, общий анализ мочи. Изменения в показателях этих анализов неспецифичны, однако они помогают выяснить, как далеко зашла болезнь, и какие органы поражены. Для подтверждения диагноза нужно выяснить содержание меди и церулоплазмина в сыворотке крови и количество меди, выделяемое с мочой за сутки. Иногда проводят генетическое исследование на наличие мутаций определенных генов.
Кроме того, делают дуплексное сканирование сосудов печени, эхокардиографию, ультразвуковое определение жидкости в брюшной полости и комплексное ультразвуковое исследование внутренних органов.
Лечение
Лечение при болезни Вильсона пожизненное. Обычно для этого используют хелатирующие соединения – вещества, образующие с медью прочные комплексы и выводящие ее из организма. Обычно это D-пеницилламин. Лечение назначается только после тщательного обследования и подбора правильной дозы этого препарата. Иногда для лечения используют соли цинка.
Если болезнь зашла далеко, необходима трансплантация печени.
При отсутствии лечения пациент погибает через 5-14 лет. Основная причина смерти – желудочно-кишечные кровотечения.
Образ жизни
Страдающие болезнью Вильсона должны отказаться от алкоголя. Они не должны принимать препараты, токсичные для печени. Важно соблюдать диету, врачи назначают им стол 5а, и ограничивать поступление меди в организм. Из рациона следует исключить продукты с высоким содержанием меди в организме:
- баранину;
- свинину;
- печень, почки;
- морепродукты;
- орехи;
- грибы;
- картофель;
- шоколад;
- какао;
- ржаной хлеб;
- минеральную воду.
Нельзя готовить пищу в медной посуде.
Профилактика
Единственная возможная профилактика болезни Вильсона – генетическое консультирование родителей. Эту процедуру стоит пройти всем родственникам страдающего этим заболеванием.
Во всех случаях необходимо назначение также специальной диеты с ограничением меди. В обязательном порядке из рациона должны быть исключены огурцы, сухофрукты, орехи, продукты из печени животных и птиц. Ограничение накладываются на употреблении шоколада, некоторых морепродуктов.
Симптомы
Заболевание, как правило, начинает развиваться в подростковом или юношеском возрасте. На первый план выходят такие симптомы, как нарастающая мышечная ригидность, гиперкинезы различной формы и амплитуды, дрожание конечностей, туловища, головы, нарушениями речи и психики. Иногда дополнительно имеют место эпилептиформные приступы. При общем осмотре нередко можно обнаружить болезненность и увеличение печени.
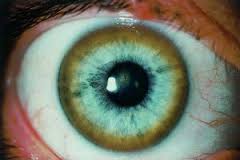
Также характерным симптомом, являющимся одним из главных критериев диагностики, является наличие зеленоватого кольца на радужке – кольцо Кайзера-Флейшера.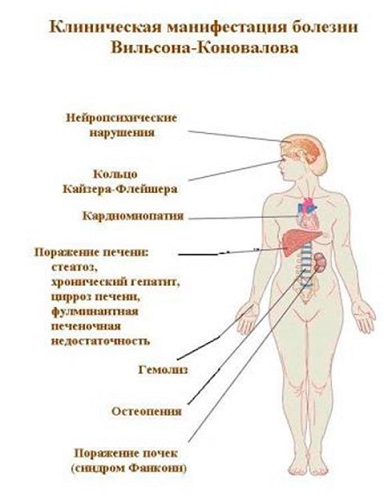
В зависимости от выраженности тех или иных симптомов выделяют различные формы заболевания (по Н.В. Коновалову):
Заболевание наследуется по аутосомно-рецессивному типу. Ген расположен в длинном плече хромосомы 13, экспрессируется в печени, почках, плаценте. Его продукт — катион, транспортирующий Р-тип АТФазного протеина (АТР7В), локализуется в цитоплазме и аппарате Гольджи клеток печени и головного мозга. Функционально важными являются.
Патогенез (что происходит?) во время Гепатолентикулярной дегенерации (болезни Вестфаля-Вильсона-Коновалова):
Известно около сотни различных мутаций, затрагивающих ген, котрый вызвает заболевание. Наиболее распространенной мутацией является H1069Q у гетерозиготных носителей гена заболевания не развивается, хотя при лабораторном исследовании выявляются субклинические изменения в метаболизме меди.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Если Вами ранее были выполнены какие-либо исследования, обязательно возьмите их результаты на консультацию к врачу. Если исследования выполнены не были, мы сделаем все необходимое в нашей клинике или у наших коллег в других клиниках.
Если Вы хотите задать вопрос врачу – воспользуйтесь разделом онлайн консультации, возможно Вы найдете там ответы на свои вопросы и прочитаете советы по уходу за собой. Если Вас интересуют отзывы о клиниках и врачах – попробуйте найти нужную Вам информацию в разделе Вся медицина. Также зарегистрируйтесь на медицинском портале Euro lab, чтобы быть постоянно в курсе последних новостей и обновлений информации на сайте, которые будут автоматически высылаться Вам на почту.
Из-за дефекта гена нарушается синтез белков, осуществляющих функцию транспорта меди. В норме медь, попадающую в организм с продуктами питания, обезвреживают печеночные клетки. Дальше она выводится в желчь и покидает организм естественным путем. При болезни Вильсона-Коновалова этот механизм нарушается. На первом этапе развития медь накапливается в гепатоцитах (клетках печени), вызывая токсический гепатит.
Классификация и симптомы болезни Вильсона-Коновалова
В России принята классификация заболевания по клиническим формам:

Болезнь может возникнуть в любом возрасте, но чаще всего ее дебют приходится на 11–25 лет. Чем позже появляются симптомы, тем медленней развивается заболевание. Описанные в литературе формы редко встречаются «в чистом» виде. Как правило, отмечаются симптомы со стороны практически всех органов и систем.
Со стороны печени болезнь Вильсона-Коновалова обычно проявляется в виде острого или хронического гепатита с исходом в цирроз печени и портальную гипертензию:
- желтуха;
- повышение температуры;
- тошнота, рвота;
- боль и тяжесть в правом подреберье;
- увеличение печени;
- на поздних стадиях — отеки, асцит, кровотечения (проявления цирроза).
Со стороны нервной системы отмечаются:
- тремор (дрожание) конечностей;
- нарушения походки;
- повышенное слюноотделение;
- маскообразное лицо;
- эпилептические приступы;
- ригидность («застывание») мышц — больные нередко замирают в самых причудливых позах.
Со стороны психики:
- аффективные вспышки;
- снижение интеллекта;
- изменения личности, «дурашливость»;
- галлюцинации, психозы.
Со стороны системы крови:
- гемолиз (распад эритроцитов);
- анемия;
- тромбоцитопения;
- нарушения свертываемости крови.
Со стороны почек:
- повышение уровня креатинина крови;
- отеки;
- микрогематурия (скрытая кровь в моче);
- протеинурия;
- нефролитиаз (камни).
Со стороны эндокринной системы:
- задержка полового созревания;
- гинекомастия;
- гирсутизм;
- ожирение;
- аменорея;
- спонтанные аборты.
Таким образом, клиническая картина складывается сложная и крайне разнообразная, что значительно затрудняет диагностику заболевания.
БВ – болезнь Вильсона
1. Краткая информация
Болезнь Вильсона (БВ) относится к числу наиболее трудно диагностируемых заболеваний печени в связи с длительным латентным течением, особенно на начальных стадиях заболевания, и большим полиморфизмом клинической симптоматики. В связи с этим болезнь Вильсона необходимо исключать у каждого пациента детского и подросткового возраста с патологией печени неуточненной этиологии.
Своевременное назначение патогенетической терапии при болезни Вильсона у детей сопровождается регрессом клинической симптоматики, предотвращением формирования цирроза печени и неврологической симптоматики, улучшением качества жизни и социальной адаптации ребенка. В связи с этим важнейшей медицинской и социальной задачей является ранняя диагностика и адекватная терапия БВ [1-10].
Болезнь Вильсона (синонимы: болезнь Вильсона-Коновалова, гепатолентикулярная дегенерация, гепатоцеребральная дистрофия) – редкое наследственное заболевание, связанное с нарушением метаболизма меди и избыточным ее накоплением в различных органах и тканях, преимущественно проявляющееся симптоматикой поражения печени и центральной нервной системы [1-10].