- низкой концентрации в сыворотке крови ионов Na, Mg, K, Cl;
- повышенном содержании в моче ионов Na, Mg, K, Cl;
- гиперфосфатемии;
- гиперкальциурии;
- отсутствии гипертензии (артериальной);
- увеличении в плазме крови уровня альдостерона и ренина;
- экскреции калликреина и простагландинов.
Синдром Барттера — заболевание, которое передается на генетическом уровне и проявляется в виде гипокалиемии (нарушение электролитных обменных процессов), гиповолемии, метаболического алкалоза (сбой кислотно-щелочного равновесия), вторичного гиперальдостеронизма и компенсаторной гиперплазии юкстагломерулярного отдела почек.
- Причины синдрома Барттера
- Симптомы синдрома Барттера
- Диагностика синдрома Барттера
- Лечение синдрома Барттера
- Прогноз синдрома Барттера
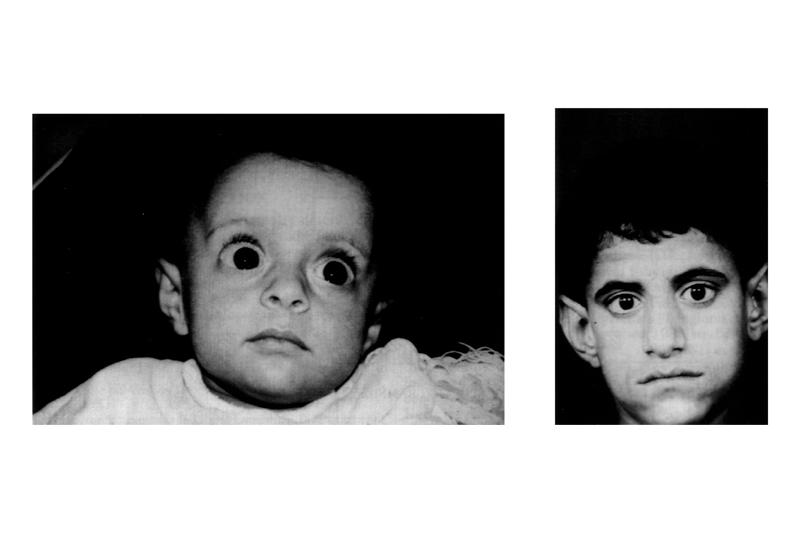
Синдором Барттера проявляется с самого рождения, а при ухудшении состояния наблюдается развитие нефрокальциноза, что в итоге приводит к почечной недостаточности. Обнаружить данное заболевание можно по следующим признакам:
- нарушение психомоторного развития у ребенка;
- гипотония мышц;
- полиурия;
- в ходе анализа результатов мочи и крови.
В ходе урологических исследований специалисты заметили, что синдром Барттера является генной мутацией дефекта петли Генле. Это в своем роде наследственность согласно аутосомно-рецессивному типу. Почечные нефроны при синдроме Баттера не способны удерживать калий, поэтому он быстро выводится из организма вместе с мочой, а циркуляция крови при этом значительно уменьшается в объеме как при пониженном, так и при нормальном АД.
Синдром Барттера классифицируют по видам согласно пораженным генам, в зависимости от характеристик их делят на:
- 1-й тип неонатального синдрома Барттера;
- 2-й тип неонатального синдрома Барттера;
- 3-й тип — классическая форма заболевания;
- 4-й тип с нейросенсорной тугоухостью;
- 5-й тип — подобный аутосомно-доминантной гипокальциемии;
- 6-й тип — синдром Гительмана (сопровождается гипомагниемией и гипокальциурием).
Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Врожденные нарушения транспорта в почечных канальцах формируют спектр редких состояний, при каждом из которых поражаются определенные сегменты нефрона. Успехи генетики и молекулярной биологии позволили расшифровать патогенез многих таких заболеваний и углубили наши представления о регуляции водно-электролитного обмена в норме.
Синдром Бартера редкая форма гипокалиемического метаболического алкалоза с гиперкальциурией наследуется аутосомно-рецессивно. Различают два клинических подтипа синдрома Бартера. Антенатальный синдром Бартера (называемый также синдромом гиперпродукции простагландина Е) обычно проявляется у новорожденных и протекает тяжелее, чем классический синдром Бартера; он включает многоводие в анамнезе, потерю соли и выраженное обезвоживание.
Более легкий классический фенотип проявляется позднее задержкой развития ребенка и частыми эпизодами обезвоживания в анамнезе. Фенотипически сходный синдром Гительмана обусловлен другим генетическим дефектом. Описан также вариант антенатального синдрома Бартера с нейросенсорной глухотой и ХПН, имеющий другую генетическую основу.
Патогенез синдрома Бартера у детей. Биохимические сдвиги при синдроме Бартера (гипокалиемический метаболический алкалоз с гиперкальциурией) напоминают последствия применения петлевых диуретиков и отражают нарушение транспорта натрия, хлорида и калия в восходящем отделе петли Генле. Потеря натрия и хлорида, приводящая к уменьшению внутрисосудистого объема, стимулирует ренин-ангиотензин-альдостероновую систему.
Альдостерон усиливает реабсорбцию натрия и секрецию калия, тем самым усугубляя гипокалиемию. Он усиливает также секрецию ионов водорода в дистальных отделах нефрона, что усугубляет метаболический алкалоз. Гипокалиемия стимулирует синтез простагландинов, которые еще больше активируют ренин-ангиотензин-альдостероновую систему. В основе синдрома Бартера лежат три разных генетических дефекта транспортеров, функционирующих на уровне петли Генле.
Каждый из них тем или иным образом участвует в транспорте натрия и хлорида. При антенатальном синдроме Бартера обнаруживаются мутации гена, кодирующего натрий/калий/2 хлоридный транспортер NKCC2 (объект действия фуросемида), или гена люминальных калиевых каналов (ROMK), тогда как для классического синдрома характерны дефекты базолатеральных хлоридных каналов (ClC-Kb).
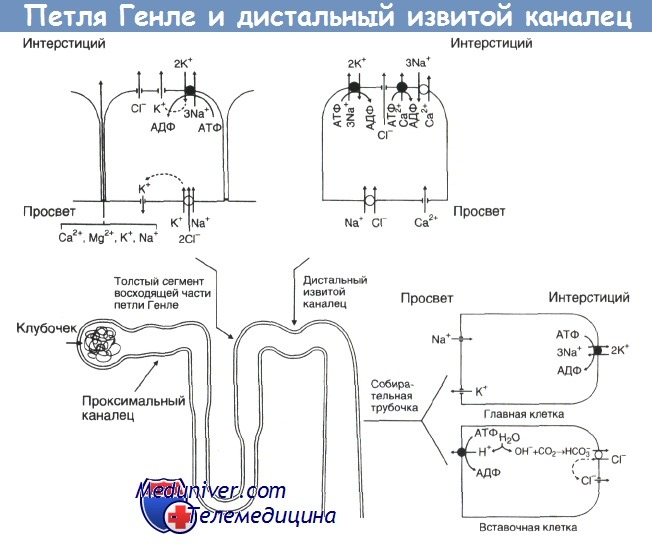 Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Содержание кальция в моче обычно повышено. Часто значительно повышен уровень ренина, альдостерона и простагландина Е в сыворотке крови, особенно при тяжелой антенатальной форме синдрома. АД в большинстве случаев нормальное, хотя обезвоживание при выраженной потере соли у больных с антенатальной формой синдрома может приводить к артериальной гипотонии. Функция почек, как правило, сохранена. При УЗИ иногда обнаруживается нефрокальциноз как следствие гиперкальциурии.
Аналогичная клиническая картина имеет место при хронической рвоте, но при этом содержание хлорида в моче снижено, тогда как при синдроме Бартера оно повышено. При гистологическом исследовании почек находят гиперплазию юкстагломерулярного аппарата. Однако диагностическая биопсия при синдроме Бартера выполняется редко.
Лечение и прогноз синдрома Бартера у детей. Терапия синдрома Бартнера направлена на предотвращение обезвоживания и поддержание питания и коррекцию гипокалиемии. Зачастую требуются очень большие дозы калия, но и в этих случаях его уровень в сыворотке не всегда удается нормализовать, особенно у новорожденных. Грудные и маленькие дети могут нуждаться и в добавках натрия. Эффективен также индометацин, ингибирующий синтез простагландинов.
При внимательном отношении к электролитному балансу, объемному статусу и росту ребенка долговременный прогноз обычно благоприятный. Однако хроническая гипокалиемия, нефрокальциноз, длительное введение индометацина могут иногда приводить к развитию интерстициального нефрита и ХПН.
Симптомы синдрома Барттера
Симптомы синдрома Барттера
Синдром Барттера проявляется или сразу после рождения, или в раннем детском возрасте. Его клиническая картина обусловлена имеющимся хроническим дефицитом калия.
При синдроме Барттера наблюдается полиурия и, как следствие, эксикоз (обезвоживание), поражение мышечной системы (слабость скелетных мышц, сердечной мышцы, гладкой мускулатуры, вялый псевдопаралич, судороги), отставание ребенка в умственном и физическом развитии, поражение нервной системы (парестезии и ригидность конечностей) при отсутствии артериальной гипертензии (нормальном или сниженном АД).
Неонатальный синдром Барттера проявляется в период внутриутробного развития плода многоводием, часто сопровождается преждевременными родами и имеет тяжелое течение. У недоношенных новорожденных наблюдается плохой аппетит, сонливость, быстрая потеря веса, задержка психомоторного развития, мышечная гипотония, нарушения зрения и слуха, гипертермия.
Классический синдром Барттера проявляется в раннем детском возрасте (после 1 года жизни) задержкой роста и развития ребенка, полиурией, склонностью к дегидратации, рвотой, запорами, полидипсией. Синдром Гительмана манифестирует примерно с 6-летнего возраста или позднее; характеризуется мышечной слабостью, утомляемостью, случаями возвратной тетании и имеет более доброкачественное течение.
При синдроме псевдо-Барттера развиваются аналогичные симптомы, обусловленные гипокалиемическим метаболическим алкалозом; данная патология часто встречается у молодых девушек, использующих для похудания диуретики и строго ограниченную диету.
Лечение синдрома Барттера
Традиционное лечение различных типов синдрома Барттера включает заместительную и медикаментозную терапию.
Необходимо обеспечение достаточного поступления калия и хлорида натрия с пищей, дополнительный прием препаратов калия. В лечении неонатального синдрома Барттера сразу же после рождения ребенка начинают экстренную интенсивную заместительную терапию с помощью инфузий солевых растворов (NaCl, KCl). Для уменьшения потери калия организмом назначают калийсберегающие диуретики (спиронолактон, триамтерен, амилорид).
Необходим прием ингибиторов синтеза простагландинов (НПВС: индометацина, аспирина) и ингибиторов АПФ (каптоприла), снижающих секрецию ренина и альдостерона. У недоношенных младенцев из-за побочного действия индометацина его применение необходимо отстрочить до достижения детьми 4-6 недельного возраста. Коррекцию гипомагниемии при синдроме Гительмана проводят препаратами магния.
Для лечения синдрома псевдо-Барттера необходимо устранить первопричину заболевания.
Никакая информация, размещенная на этой или любой другой странице нашего сайта, не может служить заменой личного обращения к специалисту. Информация не должна использоваться для самолечения и приведена только для ознакомления.
Специфические генетические мутации относятся к пяти основным типам синдрома Барттера:
Диагностика
Синдром Барттера диагностируется на основе исследования симптомов и истории болезни, а также различных анализов крови и мочи для выявления дисбаланса в уровнях соли, калия, кальция и других электролитов. Поскольку расстройство настолько редкое, почти всегда будет необходим вклад генетика, генетического консультанта и других специалистов.
Анализы крови, используемые для диагностики синдрома Барттера, позволяют выявить низкий уровень калия, хлорида, магния и бикарбоната в крови, а также повышенный уровень гормонов ренина и альдостерона.
Анализ мочи будет использоваться для выявления аномально высокого уровня натрия, хлорида, калия, кальция и магния в моче, а также наличия простагландина Е2 (маркера воспаления почек).
Антенатальные формы синдрома Барттера часто можно диагностировать до рождения, когда полигидрамниоз обнаруживается без наличия врожденных врожденных дефектов. Тут также будет повышенный уровень хлорида и альдостерона в амниотической жидкости.
Молекулярно-генетическое исследование может подтвердить диагноз. Есть несколько генетических тестов, которые могут обнаружить различные мутации, связанные с синдромом Барттера, доступные только через специализированные генетические лаборатории.
Может потребоваться дополнительное генетическое тестирование, чтобы дифференцировать синдром Барттера от тесно связанного, но более легкого наследственного расстройства, известного как синдром Гительмана.
Фуросемид-, буметанидчувствительный Na + -, K + -, 2С1-транспортный белок восходящего колена петли Генле
Код по МКБ-10
Синдром Барттера представляет собой генетически детерминированное заболевание, проявляющееся гипокалиемией, метаболическим алкалозом, гиперурикемией и повышением активности ренина и альдостерона.
Отдельно выделяют вариант Гительмана: помимо названных признаков, отмечают также гипомагниемию и гипокальциурию.
В настоящее время расшифрованы некоторые генетические механизмы синдрома Барттера и варианта Гительмана. Синдром Барттера наследуется по аутосомно-рецессивному, синдром Лиддла — по аутосомно-доминантному типу. Идентифицированы мутации, ответственные за развитие синдрома Лиддла (16р12.2-13.11 и 12р13.1).
Варианты синдрома Барттера
Тип I (неонатальный)
Фуросемид-, буметанидчувствительный Na + -, K + -, 2С1-транспортный белок восходящего колена петли Генле
АТФ-зависимый белок калиевого канала
Тиазидчувствительный транспортёр Na + и С1
[6], [7], [8], [9], [10], [11], [12]
Диагноз синдрома Барттера обычно устанавливается детским урологом по клинической симптоматике — сочетанию полиурии с мышечной гипотонией. К лабораторно-диагностическим критериям можно отнести низкую концентрацию ионов K, Cl, Na, Mg в сыворотке крови и их повышенное содержание в моче, гиперкальциурию, гиперфосфатемию, а также значительный уровень ренина и альдостерона плазмы крови, усиленную экскрецию простагландинов и калликреина с мочой, отсутствие артериальной гипертензии.
Диагностика
Диагноз синдрома Барттера обычно устанавливается детским урологом по клинической симптоматике — сочетанию полиурии с мышечной гипотонией. К лабораторно-диагностическим критериям можно отнести низкую концентрацию ионов K, Cl, Na, Mg в сыворотке крови и их повышенное содержание в моче, гиперкальциурию, гиперфосфатемию, а также значительный уровень ренина и альдостерона плазмы крови, усиленную экскрецию простагландинов и калликреина с мочой, отсутствие артериальной гипертензии.
Неонатальный тип синдрома на первой неделе жизни можно определить по наличию метаболического алкалоза с гипокалиемией, низкому удельному весу мочи, содержащей большое количество ионов K, Na, Cl, Ca, высокому уровню простагландинов в крови и моче, большой активности ренина и альдостерона в крови.
При классическом варианте течения выявляют гипокалиемический метаболический алкалоз с повышенным или нормальным содержанием кальция, не нарушенную способность концентрировать мочу. В случае синдрома Гительмана обнаруживается резко выраженная гипомагниемия и гипокальциурия. По этим показателям синдром Барттера диагностируется при исключении приема диуретиков и слабительных средств, потерь калия и хлоридов через ЖКТ.
В редких случаях возможно выполнение биопсии почки, которая позволяет выявить гиперплазию околоклубочкового аппарата. Патологию следует дифференцировать от хронической рвоты, злоупотребления мочегонными препаратами, состояний, связанных с дефицитом магния, изолированного гиперальдостеронизма, хронической надпочечниковой недостаточности.
- предотвращение потери магния и калия, употребление пищевых добавок с этими минералами
- прием препаратов (Спиронолактон, Триамтерен, Индометацин, Каптоприл, Амилорид
- прием гормона роста.
Синдром Конна — симптомы, лечение
Синдром Конна — это синоним первичного гиперальдостеронизма. Синдром Конна важно диагностировать, поскольку именно он зачастую является основной причиной высокого давления (гипертонии).
Синдром Конна — редкое заболевание, которое встречается у одного из миллиона больных. Однако это состояние фиксируется примерно у 15% больных артериальной гипертензией.
Существует две основных причинысиндрома Конна: это переизбыток альдостерона или гиперплазия (увеличение) надпочечников.
Симптомы синдрома Конна
Основные симптомы синдрома Конна:
- мышечная слабость;
- полиурия (большое количество мочи);
- никтурия (выделение большого количества мочи в ночное время);
- низкий уровень калия в крови;
- высокое давление (более 160/110 mm Hg).
Наиболее оптимальным методом диагностики является измерение в крови уровня двух гормонов: альдостерона и ренина (последний важен для выработки альдостерона).
Однако высокий уровень ренина может быть и следствием приема некоторых лекарств, например, бета-блокаторов, которыми обычно лечат артериальную гипертензию
У всех больных, чей уровень кровяного давления более 140/90, берут пробы на альдостерон и активность ренина плазмы. Образцы крови берутся при определенных условиях: утром, около 9 часов, после нахождения в лежачем положении не менее чем в течение 30 минут. Кроме того, такие пациенты нуждаются в правильном питании, соблюдении режима дня. Низкосолевая диета может некоторым образом улучшить состояние больных.
Лечение синдрома Конна
Больным с низким уровнем калия в моче может быть рекомендован дополнительный прием калия, при критически низком показателе калия пациентов помещают в стационар для лечения.
Все пациенты с подозрением на синдром Конна проходят КТ надпочечников. Этот метод диагностики выявляет причину избыточной активности этих желез: опухоль это или дисплазия.
Радикальный метод лечения — адреналэктомия, то есть удаление надпочечников, или одного из них. При двустороннем синдроме Конна удаляются оба надпочечника, при одностороннем — один. При двусторонней гиперплазии больным показан прием Спиронолактона. Это препарат, похожий по своей химической структуре на женский половой гормон эстрадиол. Альтернативный препарат: Эплеренон (Инспра). Он так же блокирует действие альдостерона, как и Спиронолактон.
Без приема лекарств или хирургического вмешательства высокий уровень кровяного давления у людей с синдромом Конна крайне тяжело контролировать. Игнорирование этого заболевания увеличивает риск инсульта, сердечно-сосудистых нарушений и почечной недостаточности.
Вегетативная нервная система (которая регулирует функции внутренних органов и некоторых мышц тела) также может быть нарушена, вызывая изменения частоты сердечных сокращений, артериального давления, опорожнения или потоотделения. Иногда СГБ приводит к затруднениям секреции в полости рта и горла. Помимо удушья и / или слюнотечения выделения могут попадать в дыхательные пути и вызывать пневмонию.
Синдром Гийена-Барре — диагностика
Начальные признаки и симптомы СГБ различны, и существует несколько расстройств с похожими симптомами. Поэтому врачам может быть сложно диагностировать СГБ на самых ранних стадиях. Врачи отмечают появление симптомов с обеих сторон тела (типичное проявление при синдроме Гийена-Барре) и скорость, с которой симптомы появляются (при других расстройствах мышечная слабость может прогрессировать в течение месяцев, а не дней или недель).
Основные диагностические результаты включают в себя:
Важный момент при лечении синдрома Туретта – это правильная обстановка дома, среди близких друзей. Человек с таким отклонением должен получать поддержку, заботу, его необходимо оградить от насмешек и неадекватных реакций на тики.
Чем лечат синдром Туретта в Москве
Все методы, которые используются при лечении подобных заболеваний, можно поделить на медикаментозные и немедикаментозные. Первые предполагают прием седативных препаратов, редко транквилизаторов. Если их и назначают, то короткими курсами и только после того, как врач выяснит, что это необходимо.

Немедикаментозные методы лечения синдрома Туретта:
Важный момент при лечении синдрома Туретта – это правильная обстановка дома, среди близких друзей. Человек с таким отклонением должен получать поддержку, заботу, его необходимо оградить от насмешек и неадекватных реакций на тики.
- полиурия, то есть частое мочеиспускание с возможностью обезвоживания;
- чрезмерная жажда;
- низкое кровяное давление;
- замедление роста;
- тенденция к развитию камней в почках;
- запор;
- усталость, мышечная слабость;
- некоторые осложнения, такие как сердечные заболевания, остеопороз.
Диагностика
Врожденный синдром Барттера диагностируется у очень маленьких детей с помощью диагностических тестов, таких как определение уровня гормонов ренина и альдостерона в крови, а также натрия, калия и кальция в моче. Также можно заметить, что ребенок набирает вес слишком медленно и плохо растет. Чтобы точно определить, какие гены являются поврежденными, может быть проведено генетическое тестирование.
Синдром Барттера следует дифференцировать от патологий с похожими симптомами: синдром Псевдобарттера, вызванный злоупотреблением диуретиками, синдром Гительмана, целиакия и муковисцидоз.
Несмотря на эти симптомы показатели артериального давления сохраняются в норме (или даже являются пониженными).
Прогноз
Синдром Барттера является генетической патологией и не может излечиваться полностью. Однако при раннем выявлении и проведении адекватного своевременного лечения во время новорожденности или в раннем возрасте специалистам удается стабилизировать состояние ребенка и уменьшать тяжесть проявлений и последствий. Например, правильно и вовремя проведенная заместительная и медикаментозная терапия позволяют смягчать проявления задержки в физическом и психическом развитии.
Тяжелое течение синдрома приводит к нефрокальцинозу, который становится причиной развития хронической почечной недостаточности. При отсутствии адекватного лечения неонатального типа патологии тяжелые электролитные нарушения и обезвоживание организма ребенка приводят к летальному исходу.