Уровень распространения синдрома делеции 22q11 (22Q11DS оценивается приблизительно как 3 случая на 10000 новорожденных (Oskarsdottir 2005) и возможно несколько чаще встречается среди девочек (личные неопубликованные данные).
Уровень распространения синдрома делеции 22q11 (22Q11DS оценивается приблизительно как 3 случая на 10000 новорожденных (Oskarsdottir 2005) и возможно несколько чаще встречается среди девочек (личные неопубликованные данные).
В настоящее время большая часть практикующих врачей рассматривает синдром делеции 22q11 как общепринятый синдром с фенотипом Ди Джорджи. Таким образом, диагноз устанавливается в случае выявления «САТСН-22 фенотипа» (аномалии сердца (Cardiac), аномалии (Anomalous) лица, гипоплазии тимуса (Thymus), расщелины (Cleft) неба (включая только подслизистое расщепление) и гипокальциемии (Hypocalcaemia) и делеции сегмента 22q11, выявленной методом FISH-диагностики.
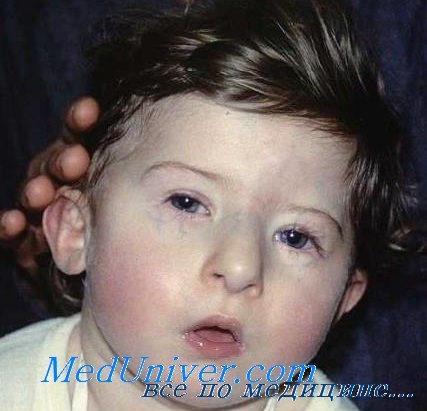
а) Клинические проявления. Тимус и паратиреоидные железы отсутствуют или эктопированы, также часто отмечается мальформация крупных сосудов основания сердца. Отсутствие тимуса может сопровождаться тяжелым иммунодефицитом, а отсутствие паращитовидных желез—тяжелыми гипокальциемическими судорогами в период новорожденности и позже. Гипокальциемия связана с паратиреоидным гормоном. Часто встречается расщелина неба, микрогнатия, низко расположенные уши и гипертелоризм (Conley et al., 1979).
Частично совпадающий фенотип, известный как вело-кардио-фациальный синдром или синдром Шпринтцена, проявляется сходными аномалиями сердца, дисморфизмом лица, расщелиной неба и задержкой умственного развития, иногда сопровождается поздним развитием психиатрических отклонений и также сочетается с делецией 22q11. Также может отмечаться гипоплазия мозжечка (Devriendt et al., 1996).
 Синдром Ди Джорджи
Синдром Ди Джорджи
Редактор: Искандер Милевски. Дата публикации: 4.12.2018
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Ди Джорджи связан с гипо- или аплазией тимуса и паращитовидных желез, приводящей к Т-клеточному иммунодефициту и гипопаратиреозу.
Синдром Ди Джорджи является результатом делеции локуса 22q11 в 22-й хромосоме, мутации генов локуса 10р13 в 10-й хромосоме и мутаций неизвестных генов, которые приводят к дизэмбриогенезу структур, развивающихся из глоточных карманов на 8 неделе гестации. Большинство случаев являются спорадическими; мальчики и девочки поражаются с одинаковой частотой. Синдром Ди Джорджи может быть частичным, при котором сохраняется функция Т-лимфоцитов, или полным, при котором функция Т-лимфоцитов полностью нарушена.
Прогноз часто зависит от тяжести пороков сердца. При частичном синдроме Ди Джорджи гипопаратиреоз лечится назначением препаратов Са и витамина D; продолжительность жизни не нарушается. При полном синдроме Ди Джорджи без лечения наступает смерть; лечение предполагает трансплантацию культуры ткани тимуса.
[1], [2], [3], [4]
Признаки и симптомы синдрома Ди Георга отличаются в зависимости от того, какой орган или система страдает и насколько серьезны дефекты. Некоторые симптомы становятся очевидными сразу после рождения, другие — могут появиться чуть позже, в раннем детстве.
Симптомы синдрома Ди Георга
Признаки и симптомы синдрома Ди Георга отличаются в зависимости от того, какой орган или система страдает и насколько серьезны дефекты. Некоторые симптомы становятся очевидными сразу после рождения, другие — могут появиться чуть позже, в раннем детстве.
Основные симптомы синдрома Ди Георга это:
Во взрослой жизни люди с синдромом Ди Георга нередко страдают от таких заболеваний, как:
- шизофрения;
- тревожные расстройства;
- депрессия.
По мере взросления у больного ребенка проявляются такие симптомы, как:
- синдром гиперактивности;
- синдром дефицита внимания;
- отсутствие социальных навыков и умения контактировать с другими детьми.
Таким детям тяжело адаптироваться в современном обществе без поддержки и заботы взрослых или общения с детьми с аналогичными расстройствами.
Расположение паращитовидных и щитовидных желез
Достаточно часто болезнь протекает в менее тяжелой форме и является наиболее распространенной причиной умственной отсталости с малым количеством других проявлений, поэтому может быть формально не диагностирована.
Для данного синдрома дисэмбриогенеза 3-4 жаберной дуги — фенотипа CATCH 22 характерно наследование по аутосомно-доминантному типу, которое наблюдается лишь в 5-10% случаев, но при этом не исключается возможность аутосомно-рецессивного типа наследования с разной экспрессивностью.
Патогенетические звенья синдрома:
Профилактика
Рекомендации специалистов, позволяющие предупредить развития тяжелых осложнений неизлечимого синдрома:
- избегать стрессов, не конфликтовать, иметь оптимистический настрой;
- не переохлаждаться, одеваться по сезону;
- не контактировать с инфекционными больными;
- беременным женщинам не употреблять алкоголь и не курить;
- своевременная вакцинация от вирусных инфекций позволяет сохранить жизнь и здоровье беременным женщинам и плоду.
Прогноз при синдроме Ди Джорджи неоднозначный. Он определяется выраженностью и степенью коррекции дисфункций сердца и эндокринных желез. В большинстве случаев он является крайне неблагоприятным. При отсутствии адекватного лечения дети погибают на первом году жизни от патологии сердца и тяжелых инфекций. Регулярное применение лекарств и особый образ жизни больных увеличивают продолжительность жизни. Но несмотря ни на что, больные дети редко доживают до 10-летнего возраста.
Синдром Ди Джорджи – это генетически детерминированный иммунодефицит, сопровождающийся аномальным строением лица и врожденными пороками сердца. Описанная патология диагностируется сразу после родов, имеет неблагоприятное течение и часто заканчивается смертью младенца. Специалисты рекомендуют будущим родителям взвесить свои моральные и материальные ресурсы прежде, чем родить больного ребенка.
Флуоресцентная гибридизация in situ (FISH) выявляет делецию локуса 22q11 в 22-й хромосоме; может быть проведено стандартное хромосомное тестирование для выявления других нарушений.
Лечение
Частичный синдром: добавки кальция и витамина D
Полный синдром: трансплантация культивированной ткани вилочковой железы или гемопоэтических стволовых клеток
При частичном синдроме Ди Джорджи гипопаратиреоз лечится препаратами, содержащими кальций, и витамином D; продолжительность жизни не нарушается.
При полном синдроме Ди Джорджи без лечения наступает смерть; лечение предполагает трансплантацию культуры ткани тимуса или гемопоэтических стволовых клеток.
Была ли страница полезной?
Положительный результат говорит о наличии в исследуемом образце микроделеции.
Генетическое исследование, позволяющее выявить причинный фактор – мутацию (отсутствие или недостаточное функционирование некоторых генов (или их частей) на 15-й отцовской хромосоме (синдром Прадера — Вилли) или мутацию материнского генетического материала аналогичного генетического региона (синдром Ангельмана, а также мутация (выпадение одного из участков) 22-й хромосомы, ведущая к гипоплазии вилочковой и паращитовидной желез (синдром Ди Джорджи).
Тест на синдромы «смежных генов», FISH-тест на генетические аномалии, FISH-анализ на Прадера – Вилли, Ангельмана, Ди Джорджи.
Синонимы английские
The test for syndromes of «adjacent genes», FISH analysis on Prader-Willy syndrome (PWS), FISH analysis for Angelman syndrome (AS), FISH analysis for Di George syndrome (DGS), FISH-test for genetic abnormalities.
Метод исследования
Дифференциальное окрашивание хромосом.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Исследование проводится в состоянии сытости, не рекомендуется сдавать кровь на данное исследование натощак.
- Исключить (по согласованию с врачом) прием антибактериальных и химиотерапевтических препаратов в течение 14 дней до исследования.
- Исследование рекомендуется проводить не ранее чем через 2 недели после перенесенных инфекционных/острых воспалительных заболеваний.
Общая информация об исследовании
Цитогенетический анализ проводится методом флуоресцентной гибридизации in situ (FISH, от англ. fluorescence in-situ hybridization), который является одним из самых современных методов диагностики хромосомных аномалий. Подробнее с методом можно ознакомиться здесь (https://helix.ru/kb/item/12-052).
Микроделеционные синдромы определяются как группа клинически узнаваемых расстройств, характеризующихся небольшой утратой хромосомного сегмента, охватывающего несколько соседних генов, каждый из которых потенциально может вносить свой вклад в фенотип. Эти синдромы называют еще синдромами «смежных генов». Большинство клинически значимых микроделеций, по-видимому, происходят спорадически, но в ряде случаев может обнаруживаться наследственная связь.
Синдром Прадера — Вилли (75% случаев) и 80% случаев возникновения синдрома Ангельмана связаны с микроделецией 15q11-13. Идентифицированы различные молекулярные механизмы, приводящие к этой потере. Примечательно, что при этом каждый из синдромов может сопровождаться и другими мелкими «молекулярными поломками».
Синдром Прадера — Вилли рассматривается как многоэтапное расстройство, характеризующееся тремя различными фазами:
FISH-анализ позволяет достоверно диагностировать общеизвестные микроделеционные синдромы и является стандартным диагностическим методом при данной патологии. Исследование позволяет установить этиологию заболевания и сделать прогноз по дальнейшему его течению.
Для чего используется исследование?
- Для диагностики микроделеций.
Когда назначается исследование?
- Пренатальная диагностика при подозрении на наличие отклонений в развитии плода.
- Постнатальная диагностика генетической патологии у ребенка при наличии соответствующих клинических признаков.
- При планировании последующих беременностей, если в семье есть ребенок с микроделеционным синдромом.
Что означают результаты?
Положительный результат говорит о наличии в исследуемом образце микроделеции.
Отрицательный результат указывает на отсутствие искомой микроделеции, но не может достоверно опровергать заболевание, так как синдром может быть вызван другим генетическим сбоем (например, однородительской дисомией, транслокацией родительских хромосом). В процентном соотношении вероятность этого значительно меньше, но все же возможна.
- встречается у 1:25000- 1:10000 новорождённых;
- с помощью обычного исследования хромосомного состава кариотипа выявить данную патологию невозможно.
- встречается примерно у 1:10000 новорождённых;
- с первых лет жизни наблюдается задержка умственного и неврологического развития.
Синдром Ди Джорджи:
- встречается у 1:3000-1:20000 новорождённых, в равной доле как у мальчиков, так и у девочек;
- характерна триада симптомов: гипоплазия тимуса и паращитовидной железы, врождённый порок сердца;
- 5-10% — аутосомно-доминантный тип наследования.
FISH-тест часто используется совместно с другими методами молекулярной и цитогенетической диагностики.
[40-006] Беременность — Пренатальный скрининг трисомий I триместра беременности (синдром Дауна) [16-001] Исследование кариотипаКто назначает исследование?
Педиатр, врач-генетик, эндокринолог, невролог.
ВВИГ — внутривенные иммуноглобулины
Определение
Синдром делеции 22-й хромосомы (синдром del 22q11) (при выраженных иммунологических изменениях синдром Ди Георга (СДГ)) — это совокупность морфологических, иммунологических и неврологических изменений, которые являются следствием делеции длинного плеча одной копии 22-й хромосомы — del 22q11.2.
В классическом понятии этот синдром представляет собой комплекс симптомов, состоящий из патологии лицевого скелета (расщелины твердого неба), врожденного порока сердца, иммунодефицита вследствие гипоплазии (аплазии) тимуса и гипокальциемии, как результат гипоплазии паращитовидной железы.
Как ни один другой синдром, синдром del 22q11.2 вариабелен в количестве признаков и степени их выраженности, что и объясняет тот факт, что исторически этот синдром в литературе имеет порядка десятка различных названий, включая синдром Ди Георга, САТСН 22, велокардиофациальный сидром, Шпринтцена синдром, Кайлера синдром, синдром лицевых и конотрункальных аномалий и т.д. Однако термин СДГ применим к случаям делеции 22q11.2 хромосомы с иммунными дефектами.
Петр, 36 лет, г. Москва
Возможные осложнения и прогноз
Самыми опасными для пациентов с синдромом Ди Джорджи являются последствия сердечной недостаточности. Они способны приводить к отеку легких и гибели ребенка. Судороги также относятся к числу осложнений при заболевании. Инфекции у малышей с недугом протекают тяжело. Возможно развитие негативных последствий со стороны дыхательной системы, пищеварительного тракта и слухового анализатора.
Исход заболевания определяется выраженностью патологических изменений. Прогноз при поражении от осторожного до неблагоприятного.
Входит в объем первичного обследования для исключения сопутствующих аномалий:
Здравствуйте!
1.2. Обнаруженная делеция, безусловно, может влиять на темпы развития. Есть данные, что нарушение развития и речи встречаются у 50%, а трудности обучения у 70% обладателей делеции.
В разделе http://forums.rusmedserv.com/forumdisplay.php?f=151 есть много полезной информации для оценки развития и возможных мер.
3.Есть много синдромов, которые похожи и имеют несколько одинаковых симптомов. Позже оказалось, что эти разные синдромы возникают в результате делеции в 22 хромосоме. Среди отличий, при синдроме ДиДжорджи есть дефект тимуса и проявление иммунодефицита. Но не обязательно. Вообще, для хромосомных аномалий свойственна значительная вариативность проявления симптомов.
4. Может быть, не это важно. Та делеция, которую определяют стандартным (коммерческим) тестом, на самом деле достаточно велика. А ваша конкретная делеция может быть меньшего размера. Но и это может не иметь значения, поскольку, как я уже говорила, почему-то проявления патологий у обладателей даже одинаковых микроделеций — могут сильно отличаться.
5. Риск аутизма самый низкий по сравнению с другими возможными проявлениями. Опять же, рекомендую раздел развития и найти подобных специалистов в реале.
Комплексное иммунологическое обследование на момент когда ребенку было 2 месяца
[Изображения доступны только зарегистрированным пользователям]
[Изображения доступны только зарегистрированным пользователям]
Исследование сывороточных иммуноглобулинов, когда ребенку было 5 месяцев
[Изображения доступны только зарегистрированным пользователям]
Последний общий анализ крови делался нами 2 месяца назад, он находится в мед карте. Наш педиатр прокомментировала его как «все в норме».
1. Что с тимусом?
2. Уровень кальция? Паратгормон определяли?
3. Загрузите протокол УЗИ почек если проводили
4. Общий анализ крови все таки постарайтесь достать.
Тяжелого комбинированного иммунодефицита нет,
а уровень T — лимфоцитов постепенно будет увеличиваться; начнут накапливаться Т-клетки памяти.
Частичная форма наиболее вероятна, но нужно еще пройтись про критериям.
Пришлите в личном сообщении фотографии лица ребенка в хорошем качестве, опубликую в скрытом режиме.
Пройдите по этой ссылке и заполните таблицы
Опубликуйте фото в теме, мы сможем сделать его доступным только для консультантов.
Проблемы со вскармливанием бывают при наличии челюстно лицевых аномалий у ребенка, как одного из симптомов.
Согласно выписки из истории болезни, протоколу проведения операции радикальной коррекции впс тимус отсутствует.
Анализ на кальций и ттг. Судорог не было и нет.
[Изображения доступны только зарегистрированным пользователям]
УЗИ почек не проводилось. Нам стоит его сделать?
Общий анализ крови достану.
Фотографию вышлю, таблицу заполню.
Входит в объем первичного обследования для исключения сопутствующих аномалий:
Initial studies — Infants with suggestive signs and symptoms should have the following performed:
●Cardiac evaluation and echocardiogram (urgently)
●Serum calcium and phosphorus levels
●Complete blood count with differential to evaluate for lymphopenia
●Chest radiograph to evaluate for absence of a thymic shadow
●Renal ultrasound to assess for anomalies
●T and B cell subsets by fluorescence-activated cell sorting (FACS)
●Immunoglobulin levels, and if appropriate, antibody responses to vaccines
| Страница 1 из 2 | 1 | 2 | > |
Работает на vBulletin® версия 3.
Copyright ©2000 — 2020, Jelsoft Enterprises Ltd.
Во время планирования беременности женщине нужно сделать комплекс противовирусных прививок.
Как и для большинства заболеваний, связанных с генетикой, специальной профилактики для синдрома Ди Джорджи не существует. Однако во избежание патологий развития плода будущей маме стоит соблюдать определенные рекомендации.
Во время планирования беременности женщине нужно сделать комплекс противовирусных прививок.
Прививка против краснухи
Краснуха не представляет серьезной опасности для взрослого человека, но может спровоцировать непоправимые нарушения в ЦНС плода.
Прививка против кори
Корь — тяжелое вирусное заболевание, представляющее угрозу для здоровья и жизни, как плода, так и матери.
Возможно, по назначению врача нужно будет сделать ещё какие-либо прививки.
- Отказаться от вредных привычек: курения, алкоголя, наркотических веществ;
- Обезопасить себя от заражения инфекциями и вирусами;
- По возможности ограничить стрессовые факторы;
- При наличии у матери частичных иммунных нарушений не стоит пренебрегать профилактической противомикробной терапией, которую может назначить лечащий врач.